Important Definitions this week
Ionisation of weak acids / bases(and the effects on their ability to cross membranes).
- Henderson hesselbalch equation describes the derivation of pH as a measure of acidity
- Ionisation of a drug markedly reduces its lipid permeability therefore reduces its ability to penetrate membranes.
- Weak acid gives H+, weak base takes H+
- With weak acids more lipid soluble at acidic pH
- With weak bases more lipid soluble at alkaline pH
- In kidneys weak acid are excreted faster if urine is alkaline and vice versa. “trapping”

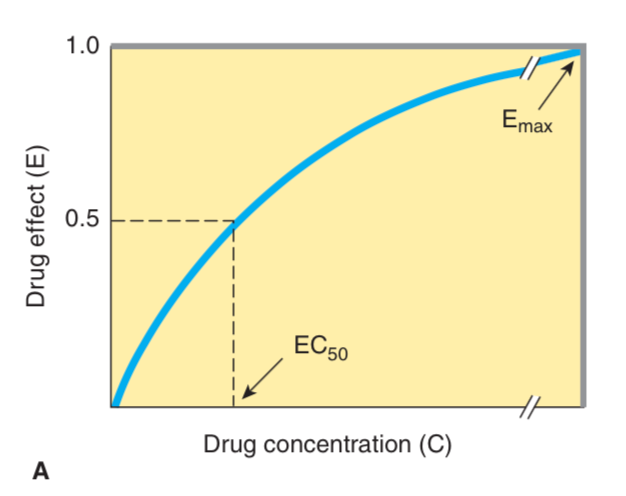
Dose – response curves.
Draw with EC50 and E max
Katzungs Basic & Clinical Pharmacology. 12th edition
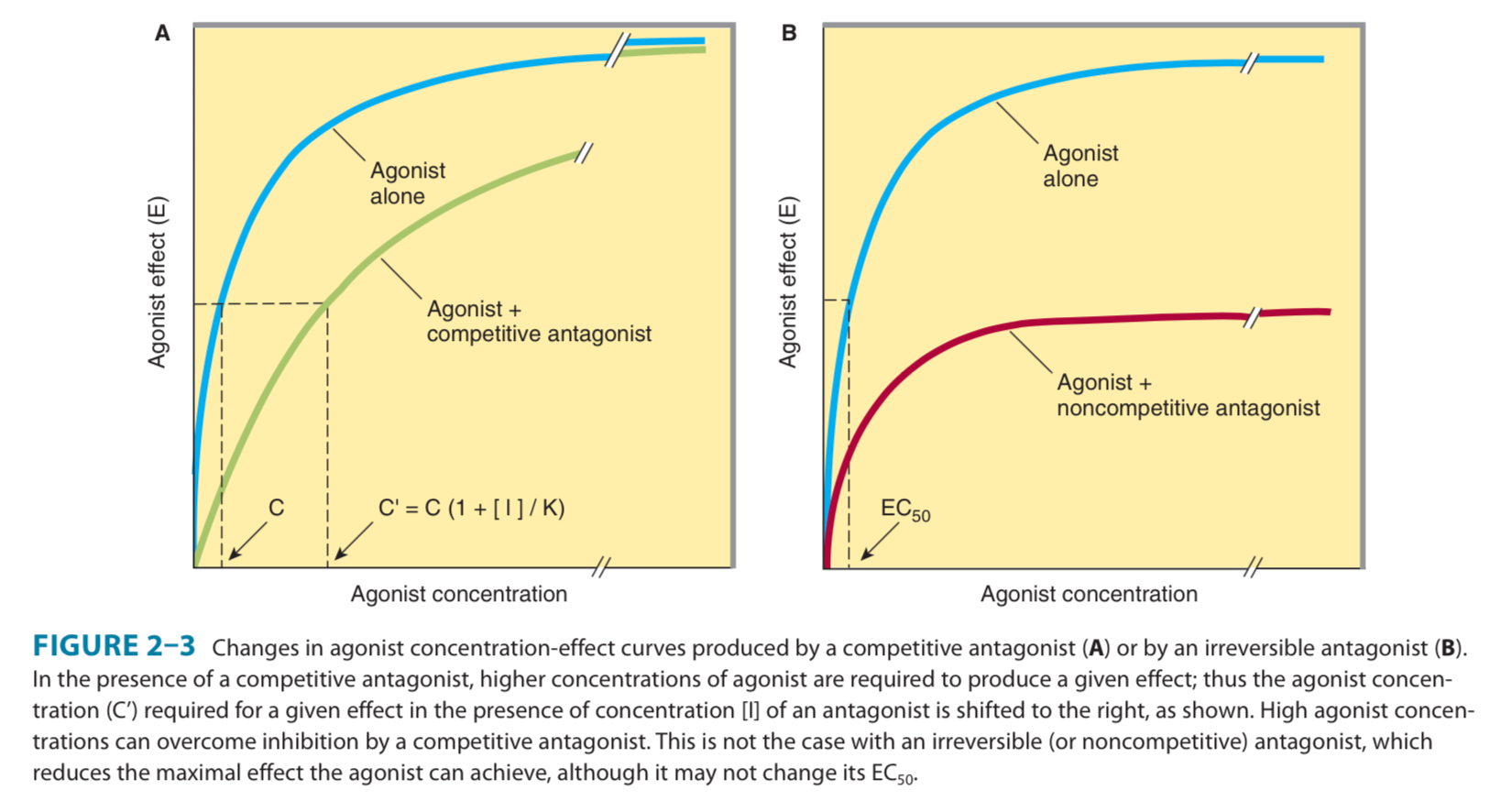
Competitive antagonist – increases EC 50 but E max is same
Irreversible antagonist – EC 50 is same but E max is reduced. (Partial agonists also have this relationship).
Katzungs Basic & Clinical Pharmacology. 12th edition
Types of antagonists.
Reversible
Irreversible
Partial agonist
Chemical antagonist
Physiological antagonist
Second messengers.
A way of transducing messages from surface to intracellular level
Ligand binds to receptor –> activation of G protein –> activation of enzyme/ion channel etc–> increase intracellular concentration of second messenger(eg Ca2+, cAMP, phosphoinositides) –> causes the biological effect of the ligand. – eg B adrenoceptors, thyrotropin receptors, glucagon
Definitions
Drug – an agent that brings about change in biological function through its chemical actions
Receptor – site where a ligand binds to effect a change or inhibit the usual action
Pharmacodynamics – actions of the drug on the body
Agonist – a molecule that binds to a receptor and activates it in some way to bring about an action either directly or indirectly.
Partial agonist – an agent that activates but to sub-maximal level
Antagonist – a molecule that binds to a receptor an inhibits its action in some way.
Potency – amount of effect for a given concentration – reflected by EC50.
Efficacy – maximal amount of effect
ED50 or EC50 – concentration required to reach 50% of maximal effect.
TD50 – median toxic dose – dose required to produce a particular toxic effect in 50% of animals.
LD50 – median lethal dose – dose required to produce a death in 50% of animals.
Therapeutic index – dose required to produce desired effect vs dose required to produce undesired effect. (TD/effective dose)
Pharmacokinetics – action of the body on the drug eg absorption, distribution(albumin binding, lipid solubility), elimination
Volume of distribution (Vd) – the apparent space in the body required to contain the drug if the drug is to be spread homogeneously at the concentration found in blood.
- Vd = drug amount/blood concentration of the drug.
- High Vd in lipophilic drugs, they have higher extravascular concetrations
Clearance – body’s ability to eliminate a drug
- Clearance = rate of elimination/plasma concentration.
- You need to add the different organ clearance together
Elimination – capacity-limited vs. flow-dependent.
First order elimination – when the elimination is not saturable, the rate of elimination is proportional to concentration. The more you give the more is cleared.
Zero order kinetics – the rate of elimination is constant regardless of concentration of the drug.
Mixed order means it behaves differently at different ranges of concentration
Most drugs when the dose is high enough elimination is saturated–> clearance will vary with concetration
Capacity limited is when blood flow to an organ doesn’t limit elimination
Rate of elimination = (Vmax x C)/(Km + C) Vmax = max elimination capacity, Km = drug concentration at which rate of elimination is 50% of max.
- Eg ethOH, phenytoin, asprin – if dose rate surpasses elimination capacity – concentration will keep increasing as long as you keep dosing.
Flow dependent elimination
- The drugs are cleared so well most of the drug is eliminated on first pass through the organ.
- Elimination of these drugs depend on the rate of drug delivery to the organ.
- Eg propanolol, verapamil
Half-life
- Time required to halve the amount of drug in the body during elimination
- T1/2 = 0.7xVd/Clearance
Bioavailability – percentage of unchanged drug reaching systemic circulation following administration by any route. Affected by absorption and first pass metabolism.
Systemic clearance is not affected by bioavailability.
First-pass elimination.
Drug metabolism in gut wall, in portal blood or most commonly Hepatic clearance of drug from portal circulation prior to reaching systemic circulation
Extraction ratio – expression of the effect of first pass metabolism on bioavailability
- ER = Liver Clearance/hepatic blood flow
Systemic bioavailability = extent of absorption x(1-ER)
Phase 1 & phase 2 reactions.
- Phase 1 = modification so a reactive and polar group is added to the drug. Can occur via oxidation, reduction, hydrolysis, cyclization
- Phase 2 = conjugation. Drug is combined with charged molecules such as glucuronidation, acetyl, sulfate, glycine. This aims to detoxify and produces more polar product–> more water soluble and easier to renally excrete
Enzyme induction & inhibition – some drugs will induce while others inhibit hepatic enzymes which alter their and other drug metabolism
- Inducers – barbiturates, carbamazepine, glucocorticoids, phenytoin, rifampicin, St Johns wort, poiglitazone
- Inhibitors – diltiazem, erythromycin, clarithromycin, fluconazole, grapefruit juice, ketaconazole
Viva questions:
- Draw a dose/response (or log dose/response) curve.
-
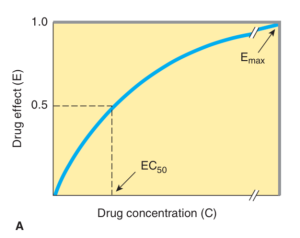
Katzungs Basic & Clinical Pharmacology. 12th edition
-
- Draw the curve after addition of a competitive antagonist / irreversible antagonist / partial agonist.
-
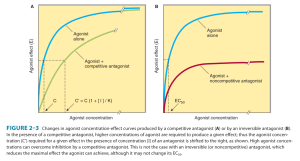
Katzungs Basic & Clinical Pharmacology. 12th edition
-
- Use the dose/response curve to demonstrate the difference between efficacy and potency. (classical viva question)
- How may drug receptors cause a response ?
- Open a channel, activate an enzyme, second messenger system–> protein production
- How do you calculate Loading dose ?
- Loading dose = Vd x target concentration
- How do you calculate Maintenance dose ?
- Dosing rate = clearance rate x target concentration
- Divide this by fraction of bioavailability
- For intermittent dose = dose rate x dose interval(hrs)
- Different dosing intervals will have same average level but different peak and trough levels.
- What is steady state ?
- A steady dose of drug remaining in the body all the time so amount of input = clearance
- Receptor effector coupling
- Receptor occupied by agonist –> conformational change –> steps –> pharmacological response.
- Spare receptors – when you can achieve maximal biological effect without occupying all the receptors (some receptors are spare). Can be spare in number or temporally be spare.
- Tachyphylaxis – respoinsiveness diminishes rapidly after administration of a drug.